PepSite
A structural method to predict peptide/protein binding
Evangelia Petsalaki, Alexander Stark, Eduardo Garcia Urdiales, Rob Russell
A structural method to predict peptide/protein binding
Evangelia Petsalaki, Alexander Stark, Eduardo Garcia Urdiales, Rob Russell
Input:
In order to perform a prediction PepSite requires:
If you think that your job is taking too long or you have any sort of problem, please contact me and I will do my best to sort out your problem.
Results:
There are two files that receive as a result of your experiment.
What do the results mean?
If you know that the peptide binds to your protein but you do not know where, PepSite can predict the correct binding site with approximately 62% sensitivity.(For more information see the Supplementary data). The lower the PEPTIDE P-VALUE the more likely it is to be correct because the signal is stronger. However even if the score is not reliable, the method performs with approximately 55% accuracy, i.e. in 55% of the cases predicted, the prediction is correct
Analysis of each individual score:
For more details concerning the specificity, the sensitivity and the accuracy of the method as well as additional validation of the method and information on how the scores are calculated please visit the Supplementary data page.
Visualization of the results
For the visualization of the results we are using Jmol, which is an open source viewer for three dimentional structures.
The options provided after PepSite runs allow you to
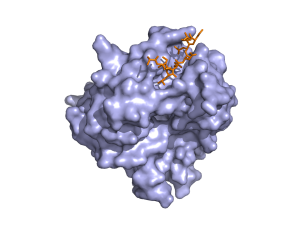
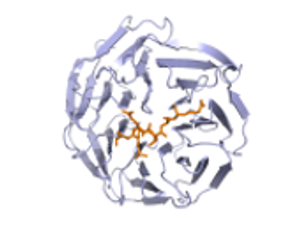
The predictions are shown in spacefilled balls and are color coded according to the residue. If you hover over the atoms/residues a label will popup. The predicted atoms are labelled as CA but in reality they are not the Ca of the amino acid but the center of mass of the active part of the residue. For information on the color codes and a quick tutorial visit this link.
For more options you can right click on the Jmol panel and there are a variety of functions to chose from. You can also chose to launch the console so that you can handle the molecules as you prefer. For details in how to use jmol, have a look at the manual pages or the Jmol Wiki
If you have any problems, questions or feedback please contact me.
In order to perform a prediction PepSite requires:
- a peptide sequence not longer than 10 amino-acid residues
- a protein structure in PDB format
If you think that your job is taking too long or you have any sort of problem, please contact me and I will do my best to sort out your problem.
Results:
There are two files that receive as a result of your experiment.
- The first file is the pdb file that contains:
- The original structure,(chain id you provided)
- The predictions for potential binding sites of each individual residue in your peptide (chain U)
- The top 9 peptides that were predicted for binding (chains 1-9).
- The second file is the predictions of the peptides, where they bind and confidence values to evaluate this.This file contains certain columns as follows:
- The first column is labeled 'Template' and shows which residues in your peptide sequence have been matched.
- The second column is labeled 'Prediction' and shows the corresponding residues in chain U that have been matched with the residues in your peptide sequence.
- The score represents the raw score for the prediction of the binding site of this peptide. It is an average of the scores of each matched residue.
- The next column is labeled 'PEPTIDE P-VALUE' and is the probability that a random match would get this score. The lower the p-value the better the prediction. The predictions are colored in the results summary table with red as the most reliable and blue the least reliable prediction.
- The next column is labeled 'RESIDUES P-VALUE' and is the average of the p-values for each of the matched residues. This value is useful for fotifying the conclusion about the peptide binding site prediction and in cases of short peptides. (details further down)
What do the results mean?
If you know that the peptide binds to your protein but you do not know where, PepSite can predict the correct binding site with approximately 62% sensitivity.(For more information see the Supplementary data). The lower the PEPTIDE P-VALUE the more likely it is to be correct because the signal is stronger. However even if the score is not reliable, the method performs with approximately 55% accuracy, i.e. in 55% of the cases predicted, the prediction is correct
Analysis of each individual score:
- PEPTIDE P-VALUE: This score represents the probability that a random match gets a score is equal or higher to this value. This means it represents how likely it is to be a random match and not a real one. The random matches were calculated by predicting the binding site for peptides included in the training dataset but not binding to the particular protein. Of course there is no guarantee that in reality they do not bind but they were treated as such. Of course the lower the PEPTIDE P-VALUE the more reliable the prediction with respect to whether it binds or not, however we consider a cutoff of 0.26 to give reasonably reliable predictions. Keep in mind that peptides smaller than 5 amino acids will tend to have much smaller scores than longer ones as they are probably even in nature more promiscuous.For this reason they will have a higher p-value, so might want to consider a higher cutoff for these cases.
- RESIDUES P-VALUE: This score shows the average p-value for each individual residue prediction. This score is included as a supplement to the PEPTIDE P-VALUE for cases that are ambiguous. For example if the peptide length is very short the PEPTIDE P-VALUE will produce a high value. To decide whether you trust the prediction or not you can have a look at the individual residue p-value and if it is low then you can trust the prediction. For these cases again a cutoff of 0.26 is recommended. However, as in some cases the specificity is very low for one residue but very high for another and this adds up to a peptide binding prediction that has a relatively high score, the PEPTIDE P-VALUE should be taken more into account if it is lower than the cutoff.
For more details concerning the specificity, the sensitivity and the accuracy of the method as well as additional validation of the method and information on how the scores are calculated please visit the Supplementary data page.
Visualization of the results
For the visualization of the results we are using Jmol, which is an open source viewer for three dimentional structures.
The options provided after PepSite runs allow you to
- select different groups of atoms and to color them by atoms, residues chain or structure.
- Also you have a variety of options to display these different groups as you prefer.
- The pull down menu that is labelled 'Predictions' shows directly each of the top 9 predicted chains and also has an option to show all the potential binding sites for each residue in your peptide (labelled 'hot spots').
The predictions are shown in spacefilled balls and are color coded according to the residue. If you hover over the atoms/residues a label will popup. The predicted atoms are labelled as CA but in reality they are not the Ca of the amino acid but the center of mass of the active part of the residue. For information on the color codes and a quick tutorial visit this link.
For more options you can right click on the Jmol panel and there are a variety of functions to chose from. You can also chose to launch the console so that you can handle the molecules as you prefer. For details in how to use jmol, have a look at the manual pages or the Jmol Wiki
If you have any problems, questions or feedback please contact me.